Source of the materials: Biopython cookbook (adapted) Status: Draft
BLAST¶
Running BLAST over the Internet
Parsing plain-text BLAST output
Hey, everybody loves BLAST right? I mean, geez, how can it get any easier to do comparisons between one of your sequences and every other sequence in the known world? But, of course, this section isn’t about how cool BLAST is, since we already know that. It is about the problem with BLAST – it can be really difficult to deal with the volume of data generated by large runs, and to automate BLAST runs in general.
Fortunately, the Biopython folks know this only too well, so they’ve developed lots of tools for dealing with BLAST and making things much easier. This section details how to use these tools and do useful things with them.
Dealing with BLAST can be split up into two steps, both of which can be done from within Biopython. Firstly, running BLAST for your query sequence(s), and getting some output. Secondly, parsing the BLAST output in Python for further analysis.
Your first introduction to running BLAST was probably via the NCBI web-service. In fact, there are lots of ways you can run BLAST, which can be categorised in several ways. The most important distinction is running BLAST locally (on your own machine), and running BLAST remotely (on another machine, typically the NCBI servers). We’re going to start this chapter by invoking the NCBI online BLAST service from within a Python script.
NOTE: The following Chapter [chapter:searchio] describes
Bio.SearchIO, an experimental module in Biopython. We intend this
to ultimately replace the older Bio.Blast module, as it provides a
more general framework handling other related sequence searching tools
as well. However, until that is declared stable, for production code
please continue to use the Bio.Blast module for dealing with NCBI
BLAST.
Running BLAST over the Internet¶
We use the function qblast() in the Bio.Blast.NCBIWWW module to
call the online version of BLAST. This has three non-optional arguments:
- The first argument is the blast program to use for the search, as a
lower case string. The options and descriptions of the programs are
available at http://www.ncbi.nlm.nih.gov/BLAST/blast_program.shtml.
Currently
qblastonly works with blastn, blastp, blastx, tblast and tblastx. - The second argument specifies the databases to search against. Again, the options for this are available on the NCBI web pages at http://www.ncbi.nlm.nih.gov/BLAST/blast_databases.shtml.
- The third argument is a string containing your query sequence. This can either be the sequence itself, the sequence in fasta format, or an identifier like a GI number.
The qblast function also take a number of other option arguments
which are basically analogous to the different parameters you can set on
the BLAST web page. We’ll just highlight a few of them here:
- The
qblastfunction can return the BLAST results in various formats, which you can choose with the optionalformat_typekeyword:"HTML","Text","ASN.1", or"XML". The default is"XML", as that is the format expected by the parser, described in section [sec:parsing-blast] below. - The argument
expectsets the expectation or e-value threshold.
For more about the optional BLAST arguments, we refer you to the NCBI’s own documentation, or that built into Biopython:
In [1]:
from Bio.Blast import NCBIWWW
help(NCBIWWW.qblast)
Help on function qblast in module Bio.Blast.NCBIWWW:
qblast(program, database, sequence, auto_format=None, composition_based_statistics=None, db_genetic_code=None, endpoints=None, entrez_query='(none)', expect=10.0, filter=None, gapcosts=None, genetic_code=None, hitlist_size=50, i_thresh=None, layout=None, lcase_mask=None, matrix_name=None, nucl_penalty=None, nucl_reward=None, other_advanced=None, perc_ident=None, phi_pattern=None, query_file=None, query_believe_defline=None, query_from=None, query_to=None, searchsp_eff=None, service=None, threshold=None, ungapped_alignment=None, word_size=None, alignments=500, alignment_view=None, descriptions=500, entrez_links_new_window=None, expect_low=None, expect_high=None, format_entrez_query=None, format_object=None, format_type='XML', ncbi_gi=None, results_file=None, show_overview=None, megablast=None)
Do a BLAST search using the QBLAST server at NCBI.
Supports all parameters of the qblast API for Put and Get.
Some useful parameters:
- program blastn, blastp, blastx, tblastn, or tblastx (lower case)
- database Which database to search against (e.g. "nr").
- sequence The sequence to search.
- ncbi_gi TRUE/FALSE whether to give 'gi' identifier.
- descriptions Number of descriptions to show. Def 500.
- alignments Number of alignments to show. Def 500.
- expect An expect value cutoff. Def 10.0.
- matrix_name Specify an alt. matrix (PAM30, PAM70, BLOSUM80, BLOSUM45).
- filter "none" turns off filtering. Default no filtering
- format_type "HTML", "Text", "ASN.1", or "XML". Def. "XML".
- entrez_query Entrez query to limit Blast search
- hitlist_size Number of hits to return. Default 50
- megablast TRUE/FALSE whether to use MEga BLAST algorithm (blastn only)
- service plain, psi, phi, rpsblast, megablast (lower case)
This function does no checking of the validity of the parameters
and passes the values to the server as is. More help is available at:
http://www.ncbi.nlm.nih.gov/BLAST/Doc/urlapi.html
Note that the default settings on the NCBI BLAST website are not quite the same as the defaults on QBLAST. If you get different results, you’ll need to check the parameters (e.g., the expectation value threshold and the gap values).
For example, if you have a nucleotide sequence you want to search against the nucleotide database (nt) using BLASTN, and you know the GI number of your query sequence, you can use:
In [2]:
from Bio.Blast import NCBIWWW
result_handle = NCBIWWW.qblast("blastn", "nt", "8332116")
---------------------------------------------------------------------------
gaierror Traceback (most recent call last)
/home/tiago_antao/miniconda/lib/python3.5/urllib/request.py in do_open(self, http_class, req, **http_conn_args)
1239 try:
-> 1240 h.request(req.get_method(), req.selector, req.data, headers)
1241 except OSError as err: # timeout error
/home/tiago_antao/miniconda/lib/python3.5/http/client.py in request(self, method, url, body, headers)
1082 """Send a complete request to the server."""
-> 1083 self._send_request(method, url, body, headers)
1084
/home/tiago_antao/miniconda/lib/python3.5/http/client.py in _send_request(self, method, url, body, headers)
1127 body = body.encode('iso-8859-1')
-> 1128 self.endheaders(body)
1129
/home/tiago_antao/miniconda/lib/python3.5/http/client.py in endheaders(self, message_body)
1078 raise CannotSendHeader()
-> 1079 self._send_output(message_body)
1080
/home/tiago_antao/miniconda/lib/python3.5/http/client.py in _send_output(self, message_body)
910
--> 911 self.send(msg)
912 if message_body is not None:
/home/tiago_antao/miniconda/lib/python3.5/http/client.py in send(self, data)
853 if self.auto_open:
--> 854 self.connect()
855 else:
/home/tiago_antao/miniconda/lib/python3.5/http/client.py in connect(self)
825 self.sock = self._create_connection(
--> 826 (self.host,self.port), self.timeout, self.source_address)
827 self.sock.setsockopt(socket.IPPROTO_TCP, socket.TCP_NODELAY, 1)
/home/tiago_antao/miniconda/lib/python3.5/socket.py in create_connection(address, timeout, source_address)
692 err = None
--> 693 for res in getaddrinfo(host, port, 0, SOCK_STREAM):
694 af, socktype, proto, canonname, sa = res
/home/tiago_antao/miniconda/lib/python3.5/socket.py in getaddrinfo(host, port, family, type, proto, flags)
731 addrlist = []
--> 732 for res in _socket.getaddrinfo(host, port, family, type, proto, flags):
733 af, socktype, proto, canonname, sa = res
gaierror: [Errno -3] Temporary failure in name resolution
During handling of the above exception, another exception occurred:
URLError Traceback (most recent call last)
<ipython-input-2-f79554f7040a> in <module>()
1 from Bio.Blast import NCBIWWW
----> 2 result_handle = NCBIWWW.qblast("blastn", "nt", "8332116")
/home/tiago_antao/miniconda/lib/python3.5/site-packages/Bio/Blast/NCBIWWW.py in qblast(program, database, sequence, auto_format, composition_based_statistics, db_genetic_code, endpoints, entrez_query, expect, filter, gapcosts, genetic_code, hitlist_size, i_thresh, layout, lcase_mask, matrix_name, nucl_penalty, nucl_reward, other_advanced, perc_ident, phi_pattern, query_file, query_believe_defline, query_from, query_to, searchsp_eff, service, threshold, ungapped_alignment, word_size, alignments, alignment_view, descriptions, entrez_links_new_window, expect_low, expect_high, format_entrez_query, format_object, format_type, ncbi_gi, results_file, show_overview, megablast)
164 message,
165 {"User-Agent": "BiopythonClient"})
--> 166 handle = _urlopen(request)
167 results = _as_string(handle.read())
168
/home/tiago_antao/miniconda/lib/python3.5/urllib/request.py in urlopen(url, data, timeout, cafile, capath, cadefault, context)
160 else:
161 opener = _opener
--> 162 return opener.open(url, data, timeout)
163
164 def install_opener(opener):
/home/tiago_antao/miniconda/lib/python3.5/urllib/request.py in open(self, fullurl, data, timeout)
463 req = meth(req)
464
--> 465 response = self._open(req, data)
466
467 # post-process response
/home/tiago_antao/miniconda/lib/python3.5/urllib/request.py in _open(self, req, data)
481 protocol = req.type
482 result = self._call_chain(self.handle_open, protocol, protocol +
--> 483 '_open', req)
484 if result:
485 return result
/home/tiago_antao/miniconda/lib/python3.5/urllib/request.py in _call_chain(self, chain, kind, meth_name, *args)
441 for handler in handlers:
442 func = getattr(handler, meth_name)
--> 443 result = func(*args)
444 if result is not None:
445 return result
/home/tiago_antao/miniconda/lib/python3.5/urllib/request.py in http_open(self, req)
1266
1267 def http_open(self, req):
-> 1268 return self.do_open(http.client.HTTPConnection, req)
1269
1270 http_request = AbstractHTTPHandler.do_request_
/home/tiago_antao/miniconda/lib/python3.5/urllib/request.py in do_open(self, http_class, req, **http_conn_args)
1240 h.request(req.get_method(), req.selector, req.data, headers)
1241 except OSError as err: # timeout error
-> 1242 raise URLError(err)
1243 r = h.getresponse()
1244 except:
URLError: <urlopen error [Errno -3] Temporary failure in name resolution>
Alternatively, if we have our query sequence already in a FASTA formatted file, we just need to open the file and read in this record as a string, and use that as the query argument:
In [ ]:
from Bio.Blast import NCBIWWW
fasta_string = open("data/m_cold.fasta").read()
result_handle = NCBIWWW.qblast("blastn", "nt", fasta_string)
We could also have read in the FASTA file as a SeqRecord and then
supplied just the sequence itself:
In [ ]:
from Bio.Blast import NCBIWWW
from Bio import SeqIO
record = SeqIO.read("data/m_cold.fasta", format="fasta")
result_handle = NCBIWWW.qblast("blastn", "nt", record.seq)
Supplying just the sequence means that BLAST will assign an identifier
for your sequence automatically. You might prefer to use the
SeqRecord object’s format method to make a FASTA string (which will
include the existing identifier):
In [ ]:
from Bio.Blast import NCBIWWW
from Bio import SeqIO
record = SeqIO.read("data/m_cold.fasta", format="fasta")
result_handle = NCBIWWW.qblast("blastn", "nt", record.format("fasta"))
This approach makes more sense if you have your sequence(s) in a
non-FASTA file format which you can extract using Bio.SeqIO (see
Chapter 5 - Sequence Input and
Output.)
Whatever arguments you give the qblast() function, you should get
back your results in a handle object (by default in XML format). The
next step would be to parse the XML output into Python objects
representing the search results (Section [sec:parsing-blast]), but you
might want to save a local copy of the output file first. I find this
especially useful when debugging my code that extracts info from the
BLAST results (because re-running the online search is slow and wastes
the NCBI computer time).
Saving blast output¶
We need to be a bit careful since we can use result_handle.read() to
read the BLAST output only once – calling result_handle.read() again
returns an empty string.
In [ ]:
with open("my_blast.xml", "w") as save_to:
save_to.write(result_handle.read())
result_handle.close()
After doing this, the results are in the file my_blast.xml and the
original handle has had all its data extracted (so we closed it).
However, the parse function of the BLAST parser (described
in [sec:parsing-blast]) takes a file-handle-like object, so we can just
open the saved file for input:
In [ ]:
with open("my_blast.xml") as result_handle:
print(result_handle)
Now that we’ve got the BLAST results back into a handle again, we are ready to do something with them, so this leads us right into the parsing section (see Section [sec:parsing-blast] below). You may want to jump ahead to that now ….
Running BLAST locally¶
Introduction¶
Running BLAST locally (as opposed to over the internet, see Section [sec:running-www-blast]) has at least major two advantages:
- Local BLAST may be faster than BLAST over the internet;
- Local BLAST allows you to make your own database to search for sequences against.
Dealing with proprietary or unpublished sequence data can be another reason to run BLAST locally. You may not be allowed to redistribute the sequences, so submitting them to the NCBI as a BLAST query would not be an option.
Unfortunately, there are some major drawbacks too – installing all the bits and getting it setup right takes some effort:
- Local BLAST requires command line tools to be installed.
- Local BLAST requires (large) BLAST databases to be setup (and potentially kept up to date).
To further confuse matters there are several different BLAST packages available, and there are also other tools which can produce imitation BLAST output files, such as BLAT.
Standalone NCBI BLAST+¶
The “new” NCBI BLAST+ suite was released in 2009. This replaces the old NCBI “legacy” BLAST package (see below).
This section will show briefly how to use these tools from within Python. If you have already read or tried the alignment tool examples in Section [sec:alignment-tools] this should all seem quite straightforward. First, we construct a command line string (as you would type in at the command line prompt if running standalone BLAST by hand). Then we can execute this command from within Python.
For example, taking a FASTA file of gene nucleotide sequences, you might want to run a BLASTX (translation) search against the non-redundant (NR) protein database. Assuming you (or your systems administrator) has downloaded and installed the NR database, you might run:
blastx -query opuntia.fasta -db nr -out opuntia.xml -evalue 0.001 -outfmt 5
This should run BLASTX against the NR database, using an expectation cut-off value of \(0.001\) and produce XML output to the specified file (which we can then parse). On my computer this takes about six minutes - a good reason to save the output to a file so you can repeat any analysis as needed.
From within Biopython we can use the NCBI BLASTX wrapper from the
Bio.Blast.Applications module to build the command line string, and
run it:
In [ ]:
from Bio.Blast.Applications import NcbiblastxCommandline
help(NcbiblastxCommandline)
In [ ]:
blastx_cline = NcbiblastxCommandline(query="data/opuntia.fasta", db="nr", evalue=0.001,
outfmt=5, out="opuntia.xml")
blastx_cline
In [ ]:
print(blastx_cline)
TODO: Need to add protein database [nr] or change example
In [ ]:
# stdout, stderr = blastx_cline()
In this example there shouldn’t be any output from BLASTX to the
terminal, so stdout and stderr should be empty. You may want to check
the output file opuntia.xml has been created.
As you may recall from earlier examples in the tutorial, the
opuntia.fasta contains seven sequences, so the BLAST XML output
should contain multiple results. Therefore use
Bio.Blast.NCBIXML.parse() to parse it as described below in
Section [sec:parsing-blast].
Other versions of BLAST¶
NCBI BLAST+ (written in C++) was first released in 2009 as a replacement
for the original NCBI “legacy” BLAST (written in C) which is no longer
being updated. There were a lot of changes – the old version had a
single core command line tool blastall which covered multiple
different BLAST search types (which are now separate commands in
BLAST+), and all the command line options were renamed. Biopython’s
wrappers for the NCBI “legacy” BLAST tools have been deprecated and will
be removed in a future release. To try to avoid confusion, we do not
cover calling these old tools from Biopython in this tutorial.
You may also come across Washington University
BLAST (WU-BLAST), and its successor,
Advanced Biocomputing BLAST (AB-BLAST,
released in 2009, not free/open source). These packages include the
command line tools wu-blastall and ab-blastall, which mimicked
blastall from the NCBI “legacy” BLAST suite. Biopython does not
currently provide wrappers for calling these tools, but should be able
to parse any NCBI compatible output from them.
Parsing BLAST output¶
As mentioned above, BLAST can generate output in various formats, such as XML, HTML, and plain text. Originally, Biopython had parsers for BLAST plain text and HTML output, as these were the only output formats offered at the time. Unfortunately, the BLAST output in these formats kept changing, each time breaking the Biopython parsers. Our HTML BLAST parser has been removed, but the plain text BLAST parser is still available (see Section [sec:parsing-blast-deprecated]). Use it at your own risk, it may or may not work, depending on which BLAST version you’re using.
As keeping up with changes in BLAST became a hopeless endeavor, especially with users running different BLAST versions, we now recommend to parse the output in XML format, which can be generated by recent versions of BLAST. Not only is the XML output more stable than the plain text and HTML output, it is also much easier to parse automatically, making Biopython a whole lot more stable.
You can get BLAST output in XML format in various ways. For the parser, it doesn’t matter how the output was generated, as long as it is in the XML format.
- You can use Biopython to run BLAST over the internet, as described in section [sec:running-www-blast].
- You can use Biopython to run BLAST locally, as described in section [sec:running-local-blast].
- You can do the BLAST search yourself on the NCBI site through your web browser, and then save the results. You need to choose XML as the format in which to receive the results, and save the final BLAST page you get (you know, the one with all of the interesting results!) to a file.
- You can also run BLAST locally without using Biopython, and save the output in a file. Again, you need to choose XML as the format in which to receive the results.
The important point is that you do not have to use Biopython scripts to
fetch the data in order to be able to parse it. Doing things in one of
these ways, you then need to get a handle to the results. In Python, a
handle is just a nice general way of describing input to any info source
so that the info can be retrieved using read() and readline()
functions (see Section sec:appendix-handles).
If you followed the code above for interacting with BLAST through a
script, then you already have result_handle, the handle to the BLAST
results. For example, using a GI number to do an online search:
In [ ]:
from Bio.Blast import NCBIWWW
result_handle = NCBIWWW.qblast("blastn", "nt", "8332116")
If instead you ran BLAST some other way, and have the BLAST output (in
XML format) in the file my_blast.xml, all you need to do is to open
the file for reading:
In [ ]:
result_handle = open("my_blast.xml", 'r')
Now that we’ve got a handle, we are ready to parse the output. The code to parse it is really quite small. If you expect a single BLAST result (i.e., you used a single query):
In [ ]:
from Bio.Blast import NCBIXML
blast_record = NCBIXML.read(result_handle)
or, if you have lots of results (i.e., multiple query sequences):
In [ ]:
from Bio.Blast import NCBIXML
blast_records = NCBIXML.parse(result_handle)
Just like Bio.SeqIO and Bio.AlignIO (see
Chapters [chapter:Bio.SeqIO] and [chapter:Bio.AlignIO]), we have a pair
of input functions, read and parse, where read is for when
you have exactly one object, and parse is an iterator for when you
can have lots of objects – but instead of getting SeqRecord or
MultipleSeqAlignment objects, we get BLAST record objects.
To be able to handle the situation where the BLAST file may be huge,
containing thousands of results, NCBIXML.parse() returns an
iterator. In plain English, an iterator allows you to step through the
BLAST output, retrieving BLAST records one by one for each BLAST search
result:
TODO: should use an example with more than one record
In [ ]:
from Bio.Blast import NCBIXML
result_handle = open("my_blast.xml", 'r')
blast_records = NCBIXML.parse(result_handle)
blast_record = next(blast_records)
print(blast_record.database_sequences)
# # ... do something with blast_record
Or, you can use a for-loop:
In [ ]:
from Bio.Blast import NCBIXML
result_handle = open("my_blast.xml", 'r')
blast_records = NCBIXML.parse(result_handle)
for blast_record in blast_records:
print(blast_record.database_sequences)
# Do something with blast_record
Note though that you can step through the BLAST records only once. Usually, from each BLAST record you would save the information that you are interested in. If you want to save all returned BLAST records, you can convert the iterator into a list:
In [ ]:
from Bio.Blast import NCBIXML
result_handle = open("my_blast.xml", 'r')
blast_records = NCBIXML.parse(result_handle)
blast_records = list(blast_records)
blast_records
Now you can access each BLAST record in the list with an index as usual. If your BLAST file is huge though, you may run into memory problems trying to save them all in a list.
Usually, you’ll be running one BLAST search at a time. Then, all you
need to do is to pick up the first (and only) BLAST record in
blast_records:
In [ ]:
from Bio.Blast import NCBIXML
result_handle = open("my_blast.xml", 'r')
blast_records = NCBIXML.parse(result_handle)
blast_record = next(blast_records)
or more elegantly:
In [ ]:
from Bio.Blast import NCBIXML
result_handle = open("my_blast.xml", 'r')
blast_records = NCBIXML.parse(result_handle)
I guess by now you’re wondering what is in a BLAST record.
The BLAST record class¶
A BLAST Record contains everything you might ever want to extract from the BLAST output. Right now we’ll just show an example of how to get some info out of the BLAST report, but if you want something in particular that is not described here, look at the info on the record class in detail, and take a gander into the code or automatically generated documentation – the docstrings have lots of good info about what is stored in each piece of information.
To continue with our example, let’s just print out some summary info about all hits in our blast report greater than a particular threshold. The following code does this:
In [ ]:
E_VALUE_THRESH = 0.04
In [ ]:
from Bio.Blast import NCBIXML
result_handle = open("my_blast.xml", 'r')
blast_records = NCBIXML.parse(result_handle)
for alignment in blast_record.alignments:
for hsp in alignment.hsps:
if hsp.expect < E_VALUE_THRESH:
print('****Alignment****')
print('sequence:', alignment.title)
print('length:', alignment.length)
print('e value:', hsp.expect)
print(hsp.query[0:75] + '...')
print(hsp.match[0:75] + '...')
print(hsp.sbjct[0:75] + '...')
Basically, you can do anything you want to with the info in the BLAST report once you have parsed it. This will, of course, depend on what you want to use it for, but hopefully this helps you get started on doing what you need to do!
An important consideration for extracting information from a BLAST
report is the type of objects that the information is stored in. In
Biopython, the parsers return Record objects, either Blast or
PSIBlast depending on what you are parsing. These objects are
defined in Bio.Blast.Record and are quite complete.
Here are my attempts at UML class diagrams for the Blast and
PSIBlast record classes. If you are good at UML and see
mistakes/improvements that can be made, please let me know. The Blast
class diagram is shown in Figure
The PSIBlast record object is similar, but has support for the rounds that are used in the iteration steps of PSIBlast. The class diagram for PSIBlast is shown in Figure.
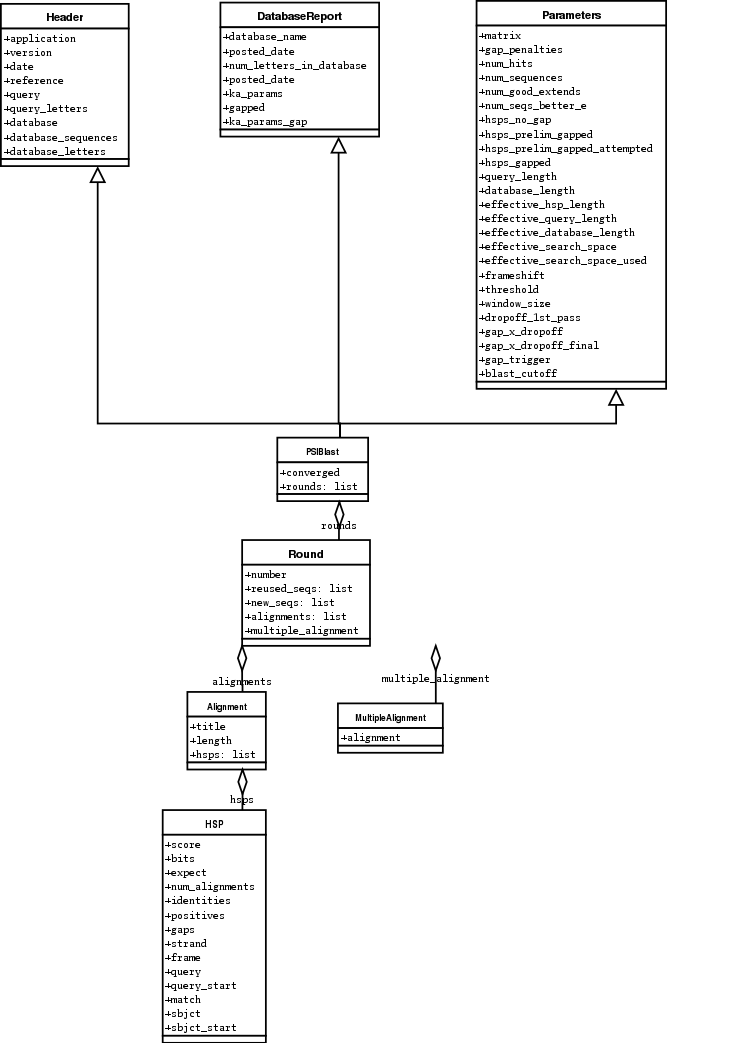
Fig. 1 Class diagram for the PSIBlast Record class.
Deprecated BLAST parsers¶
Bio.Blast.NCBIStandalone¶
The three functions for calling the “legacy” NCBI BLAST command line tools blastall, blastpgp and rpsblast were declared obsolete in Biopython Release 1.53, deprecated in Release 1.61, and removed in Release 1.64. Please use the BLAST+ wrappers in Bio.Blast.Applications instead.
The remainder of this module is a parser for the plain text BLAST output, which was declared obsolete in Release 1.54, and deprecated in Release 1.63.
For some time now, both the NCBI and Biopython have encouraged people to parse the XML output instead, however Bio.SearchIO will initially attempt to support plain text BLAST output.
Older versions of Biopython had parsers for BLAST output in plain text or HTML format. Over the years, we discovered that it is very hard to maintain these parsers in working order. Basically, any small change to the BLAST output in newly released BLAST versions tends to cause the plain text and HTML parsers to break. We therefore recommend parsing BLAST output in XML format, as described in section [sec:parsing-blast].
Depending on which BLAST versions or programs you’re using, our plain text BLAST parser may or may not work. Use it at your own risk!
Parsing plain-text BLAST output¶
The plain text BLAST parser is located in Bio.Blast.NCBIStandalone.
As with the XML parser, we need to have a handle object that we can pass
to the parser. The handle must implement the readline() method and
do this properly. The common ways to get such a handle are to either use
the provided blastall or blastpgp functions to run the local
blast, or to run a local blast via the command line, and then do
something like the following:
In [ ]:
# result_handle = open("my_file_of_blast_output.txt", 'r')
Well, now that we’ve got a handle (which we’ll call result_handle),
we are ready to parse it. This can be done with the following code:
In [ ]:
# from Bio.Blast import NCBIStandalone
# blast_parser = NCBIStandalone.BlastParser()
# blast_record = blast_parser.parse(result_handle)
This will parse the BLAST report into a Blast Record class (either a Blast or a PSIBlast record, depending on what you are parsing) so that you can extract the information from it. In our case, let’s just print out a quick summary of all of the alignments greater than some threshold value.
In [ ]:
E_VALUE_THRESH = 0.04
for alignment in blast_record.alignments:
for hsp in alignment.hsps:
if hsp.expect < E_VALUE_THRESH:
print('****Alignment****')
print('sequence:', alignment.title)
print('length:', alignment.length)
print('e value:', hsp.expect)
print(hsp.query[0:75] + '...')
print(hsp.match[0:75] + '...')
print(hsp.sbjct[0:75] + '...')
If you also read the section [sec:parsing-blast] on parsing BLAST XML output, you’ll notice that the above code is identical to what is found in that section. Once you parse something into a record class you can deal with it independent of the format of the original BLAST info you were parsing. Pretty snazzy!
Sure, parsing one record is great, but I’ve got a BLAST file with tons of records – how can I parse them all? Well, fear not, the answer lies in the very next section.
Parsing a plain-text BLAST file full of BLAST runs¶
We can do this using the blast iterator. To set up an iterator, we first set up a parser, to parse our blast reports in Blast Record objects:
In [ ]:
# from Bio.Blast import NCBIStandalone
# blast_parser = NCBIStandalone.BlastParser()
Then we will assume we have a handle to a bunch of blast records, which
we’ll call result_handle. Getting a handle is described in full
detail above in the blast parsing sections.
Now that we’ve got a parser and a handle, we are ready to set up the iterator with the following command:
In [ ]:
# blast_iterator = NCBIStandalone.Iterator(result_handle, blast_parser)
The second option, the parser, is optional. If we don’t supply a parser, then the iterator will just return the raw BLAST reports one at a time.
Now that we’ve got an iterator, we start retrieving blast records
(generated by our parser) using next():
In [ ]:
# blast_record = next(blast_iterator)
Each call to next will return a new record that we can deal with. Now we can iterate through these records and generate our old favorite, a nice little blast report:
In [ ]:
# for blast_record in blast_iterator:
# E_VALUE_THRESH = 0.04
# for alignment in blast_record.alignments:
# for hsp in alignment.hsps:
# if hsp.expect < E_VALUE_THRESH:
# print('****Alignment****')
# print('sequence:', alignment.title)
# print('length:', alignment.length)
# print('e value:', hsp.expect)
# if len(hsp.query) > 75:
# dots = '...'
# else:
# dots = ''
# print(hsp.query[0:75] + dots)
# print(hsp.match[0:75] + dots)
# print(hsp.sbjct[0:75] + dots)
The iterator allows you to deal with huge blast records without any memory problems, since things are read in one at a time. I have parsed tremendously huge files without any problems using this.
Finding a bad record somewhere in a huge plain-text BLAST file¶
One really ugly problem that happens to me is that I’ll be parsing a huge blast file for a while, and the parser will bomb out with a ValueError. This is a serious problem, since you can’t tell if the ValueError is due to a parser problem, or a problem with the BLAST. To make it even worse, you have no idea where the parse failed, so you can’t just ignore the error, since this could be ignoring an important data point.
We used to have to make a little script to get around this problem, but
the Bio.Blast module now includes a BlastErrorParser which
really helps make this easier. The BlastErrorParser works very
similar to the regular BlastParser, but it adds an extra layer of
work by catching ValueErrors that are generated by the parser, and
attempting to diagnose the errors.
Let’s take a look at using this parser – first we define the file we are going to parse and the file to write the problem reports to:
In [ ]:
# import os
# blast_file = os.path.join(os.getcwd(), "blast_out", "big_blast.out")
# error_file = os.path.join(os.getcwd(), "blast_out", "big_blast.problems")
Now we want to get a BlastErrorParser:
In [ ]:
# from Bio.Blast import NCBIStandalone
# error_handle = open(error_file, "w")
# blast_error_parser = NCBIStandalone.BlastErrorParser(error_handle)
Notice that the parser take an optional argument of a handle. If a handle is passed, then the parser will write any blast records which generate a ValueError to this handle. Otherwise, these records will not be recorded.
Now we can use the BlastErrorParser just like a regular blast
parser. Specifically, we might want to make an iterator that goes
through our blast records one at a time and parses them with the error
parser:
In [ ]:
# result_handle = open(blast_file)
# iterator = NCBIStandalone.Iterator(result_handle, blast_error_parser)
We can read these records one a time, but now we can catch and deal with errors that are due to problems with Blast (and not with the parser itself):
In [ ]:
# try:
# next_record = next(iterator)
# except NCBIStandalone.LowQualityBlastError as info:
# print("LowQualityBlastError detected in id %s" % info[1])